When studying a system’s chemical kinetics, it’s common to use perfectly mixed batch reactor assumptions and design experiments that keep mixing conditions ideal. Such assumptions include perfectly mixed (ideal tank reactors) and perfectly unmixed (ideal plug flow reactors). In reality, however, it’s rare that all of the reactor’s parts behave the same way. Space-dependent modeling is thus essential in understanding and optimizing chemical reactors. Let’s explore the development of a detailed reactor model, starting with a simple perfectly mixed example.
Our Starting Point: The Assumption of a Perfectly Mixed Batch Reactor
As a reader of the COMSOL Blog, you may already be familiar with the Chemical Kinetics series in which my colleague Eyal Spier discussed the principles of chemical kinetics. These blog posts emphasize the fundamentals of kinetic analysis, while demonstrating how you can solve trickier kinetic problems and extract quantitative data for reaction rates using the Reaction Engineering interface, available in the Chemical Reaction Engineering Module in COMSOL Multiphysics.
The first part of the Protein Adsorption tutorial in the Application Gallery offers a great example of such an analysis. In the example, we consider the performance of a reactor under the perfectly mixed assumption, using the reactor to learn more about the kinetics of the adsorption of a protein in an ion-exchange column. Later on, we add a 3D analysis, observing the variation in concentrations as a function of both space and time. The figure below illustrates how the bulk concentration of ions may vary in the real system.
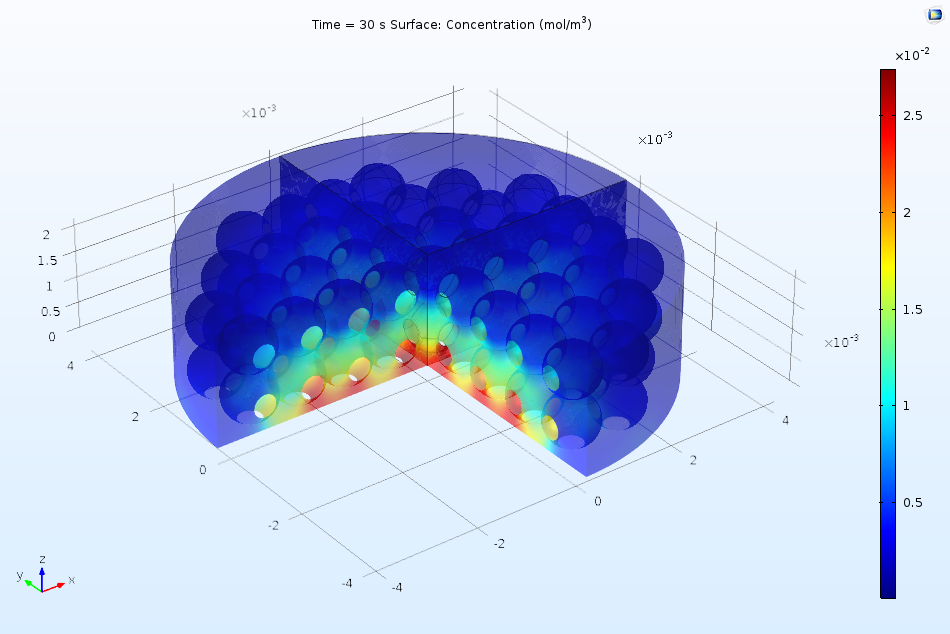
Spatial variation of ion concentration in the 3D analysis of a protein adsorption process via ion exchange.
Proteins in a solution can bind to a reactive surface, displacing smaller ions that have already been adsorbed at the surface. The ions are released into the solution, as illustrated above. We can write such a process as a chemical reaction formula:
P(aq) + nS(ads) <=> P(ads) + nS(aq)
Here, (aq) represents a chemical in a free aqueous solution, while (ads) represents a chemical bound by adsorption to a solid surface. The quantity n is the number of ions displaced by each protein molecule P. In our case, we’ll look at the behavior of two proteins, denoted A and B.
To describe a typical ion-exchange column under the perfectly mixed assumption, we consider a continuous stirred tank reactor (CSTR) at a constant volume. It is a system with a feed inlet and outlet, enabling the reactant to be continually added. As the reactant is added, the reactor volume is kept constant, meaning that the outlet flow rate must match the inlet flow rate.
In the model tree of the Reaction Engineering interface, you can see the system of reactions and the partaking chemical species. There are two surface reaction nodes that define the ion displacement reactions by proteins A and B. As such, three surface species are defined on the surface, and three additional species are defined in the free solution. The ion molarity is expressed per mole of protein, so that a unit stoichiometry of S(ads) and S appears in each ion-exchange reaction.
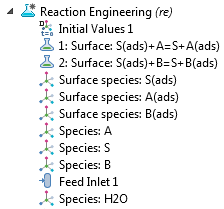
The model tree for a perfectly mixed analysis.
The solvent species water (denoted as “H2O”) is also included and is present in excess at a constant concentration. The solvent species may contribute to the analysis of the viscosity or thermal properties of the reaction mixture. These may be needed if thermal and flow analyses are performed. Take note of the feed inlet, which represents the continuing addition of a protein-containing feed to the reactor. The inlet specification uses a Gaussian pulse function in which the inlet rate is nonnegligible between approximately 0.5 seconds and 9.5 seconds after the start of the simulation.
The figure below depicts the predicted variation of the different chemical species concentrations as a function of time.
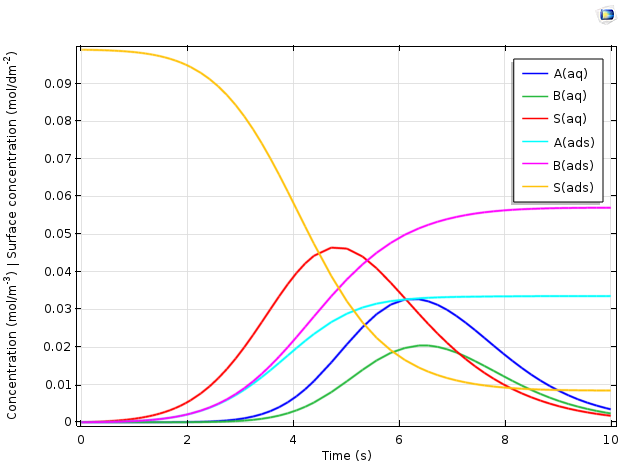
Plot comparing concentration vs. time for the adsorption of proteins A and B in an ion-exchange column.
The results above explain important phenomena in the transient behavior of the reactor. Overall, adsorbed ions S(ads), represented in yellow, are replaced with the adsorbed proteins A(ads) and B(ads). The time-dependent variation of the dissolved species concentrations is more complex. As the protein feed stream is initially added, it displaces the ions rapidly. Hence, the adsorbed protein concentrations increase and the free ion concentration in the solution rises. The dissolved concentrations A(aq) and B(aq), meanwhile, remain low, as the proteins are adsorbed rapidly and thus remain at a low concentration in the solution.
Only as the surface becomes saturated with the proteins does the rate of adsorption slow down. This is reflected by a maximum in the ion concentration in the solution, S(aq), after which the ions are removed from the column more rapidly than they are generated by the displacement from the ion-exchange surface. The concentrations of proteins in the solution now rise, as they are introduced to the reactor faster than they can react by adsorption, given the reduced density of available ion-exchange sites that remain. Finally, as the inlet concentrations of proteins tend back toward zero, the dissolved protein concentration reaches a maximum and then becomes lower as material leaves the reactor.
Using Space-Dependent Modeling to Simulate an Ion-Exchange Column for Protein Adsorption
While the CSTR analysis above can help you understand the concentration relations, it is not a completely realistic configuration. In reality, ion exchange occurs on the surfaces of a porous structure. Considering mass transport by diffusion and convection, some parts of the reactive surface are more accessible than others. To understand the real-world performance of the ion-exchange column, we must move to a 3D space-dependent analysis that can account for nonideality and mass transport.
We can retain the same chemical mechanism and move directly to a space-dependent model via the Generate Space-Dependent Model feature in the Reaction Engineering interface. As a result, a new component with a 2D or 3D geometry is automatically generated, and a chemical species transport interface is used to describe the mass balance. The reaction sink and source rates are computed through coupling to the Chemistry interface, which can be regarded as a chemistry container that stores the kinetic details, copied from the Reaction Engineering interface. It is also possible to add momentum balance (fluid flow) or energy balance (heat transfer) automatically, with properties again taken from the details of the solvent or chemical mixture in the Chemistry interface.
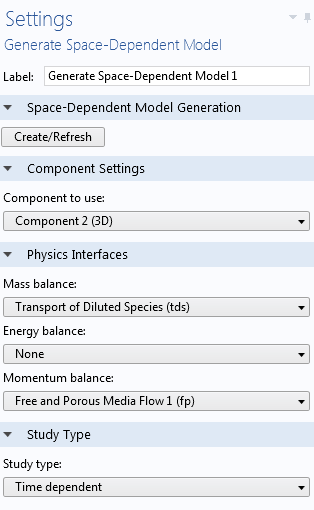
Settings for the Generate Space-Dependent Model feature.
Once the new space-dependent model component is generated, the geometry can be specified. The figure below illustrates the relation of the modeled 3D geometry to the whole reactor. While the CSTR model attempts to describe the entire reactor, the 3D analysis focuses only on a small section that is close to the inlet. To further reduce the computational size of the problem, we model only a quarter of the geometry using built-in symmetry boundary conditions to express the symmetry of the flow velocity and concentration distributions.
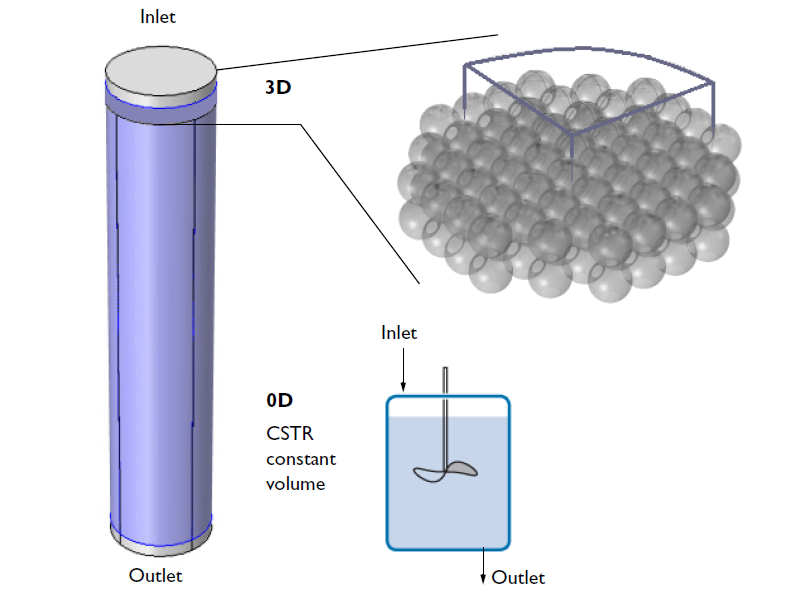
A schematic depicting the overall ion-exchange column and a small section of the porous ion-exchange beads. One quarter of the cross section is included in the 3D model.
To describe the fluid flow of water in the column, we add the Free and Porous Media Flow interface. Like the reaction kinetics, the density and viscosity of the fluid are defined by the Chemistry interface. A uniform flow velocity is defined at the outlet, representing the continuing flow along the column, downstream from the modeled region. Correspondingly, the pressure at the inlet is treated as uniform.
Let’s now look at the Transport of Diluted Species interface, which solves for mass transport by diffusion and convection. When using the Generate Space-Dependent Model feature, the automatic creation of this chemical species transport interface ensures that the correct number of chemical species are included. One concentration variable is solved for through space, accounting for each diluted chemical species considered in the CSTR problem. (They are the protein concentrations “cA” and “cB” and the free ion concentration “cS”.)
To describe the locations where the mass of different chemical species enter and leave the reactor, as well as where the specified surface reactions take place, boundary conditions are added automatically at the inlet via the Transport of Diluted Species interface. The inflow condition indicates the composition of the solution at the inlet, while the outflow condition at the top of the reactor represents the mass leaving due to fluid flow. The two surface equilibrium reaction conditions apply the necessary mass sink or source for the solution-phase species in order to describe the ion-exchange reactions of each protein.
Although the space-dependent model is created with these features ready-to-use, the 3D model in COMSOL Multiphysics cannot know which boundaries in the geometry correspond to the inlet, outlet, or reacting surfaces. The user must choose a boundary selection for each feature to correlate the mass sources and sinks from the perfectly mixed analysis to specific locations in space. The following figure depicts the nonuniform bead surface at which the ion-exchange reactions take place.
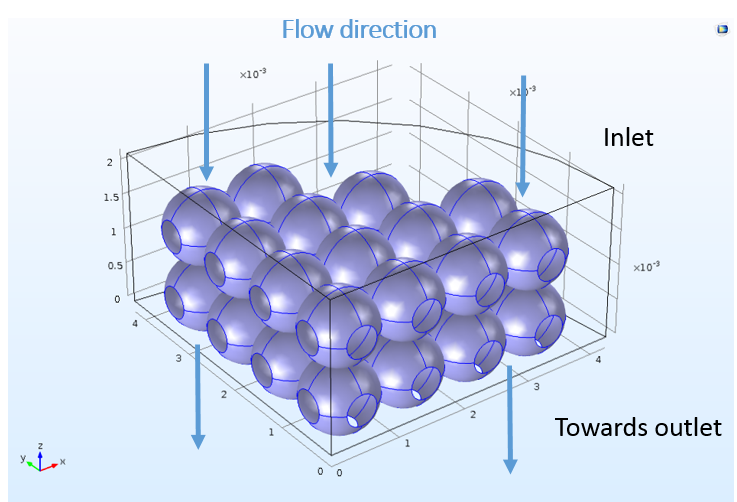
The complicated, realistic 3D bead structure used for the space-dependent mass transport analysis.
The concentration of adsorbed proteins or ions also becomes a variable across the ion-exchange resin surface. Understanding nonuniformity in the resulting protein distribution is a key output of the 3D analysis. The Surface Reactions interface solves for the surface coverage of the adsorbed species, with a mass source or sink automatically coupled to the surface reaction terms in the Transport of Diluted Species interface. Such a combination of physical equations ensures the overall conservation of mass across a combination of the bulk solution and the ion-exchange surface.
The figure below indicates the surface coverage of the adsorbed protein B(ads) after 10 seconds of exposure to the flow in the ion-exchange column. Clearly, a larger quantity of proteins adsorb to the top surface of the beads. We can attribute this to the fact that the surface is more accessible to the incident flow due to the downward direction of convection in the system. As time passes, the surface begins to saturate more quickly, meaning that future adsorption of the proteins must occur on the less accessible regions.
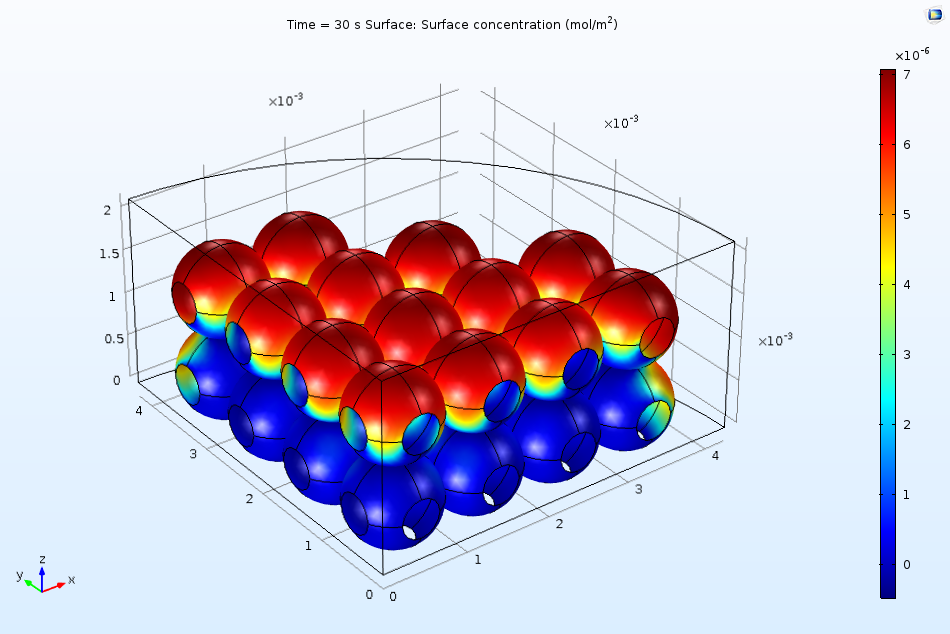
Nonuniform surface coverage of proteins after 10 seconds of exposure to an inlet stream.
Such results provide evidence for a range of timescales of responses in the ion-exchange column, where some parts of the ion-exchange surface react more quickly than others. The more complicated space-dependent models require more detail in their setup and take longer to compute. They are, however, able to provide more realistic results as well. Only space-dependent models can reveal aspects of the protein adsorption efficiency that depend on the spatial layout of the ion-exchange beads and the mass transport to their surfaces.
The various features for perfectly mixed and space-dependent modeling available in the Chemical Reaction Engineering Module are complementary, and they may become relevant at different stages of the reactor design process. Converting between 0D and 3D models is easy with the Generate Space-Dependent Model feature and the Chemistry interface, giving you the ability to copy a reaction specification and a kinetic mechanism into a space-dependent analysis.
Next Steps
To find out more, we encourage you to download the Protein Adsorption tutorial from our Application Gallery.


Comments (3)
Christian BERDUCAT
June 18, 2019Dear Edmund,
Is it possible to use Reactive Pellet Bed? I would like to simulate an Ion Exchange Resin. In this case we have the capacity of the resin instead of the surface capacity. It is a model named Nernst Model. I think you know this very well. Could you help me? I don’t see how to proceed.
Best Regards
Christian Berducat
Brianne Christopher
June 18, 2019 COMSOL EmployeeHello Christian,
Thank you for your comment.
For questions related to your modeling, please contact our Support team.
Online Support Center: https://www.comsol.com/support
Email: support@comsol.com
Kelsey Levine
July 14, 2019Does the space-dependent model generated from Reaction Engineering automatically couple the Chemistry and Transport of Diluted Species interfaces?